biocmask provides efficient abstractions to the SummarizedExperiment such that using common dplyr functions feels as natural to operating on a data.frame or tibble. biocmask was built as an alternative to the tidySummarizedExperiment package but there may be a future in which their conflicts are reconciled. biocmask uses data-masking from the rlang package in order to connect dplyr functions to SummarizedExperiment slots in a manner that aims to be intuitive and avoiding ambiguity in outcomes.
Note: This package is still under active development as of Fall 2024.
data masking SummarizedExperiment
The SummarizedExperiment object contains three main components/“contexts” that we mask, the assays(), rowData()1 and colData().
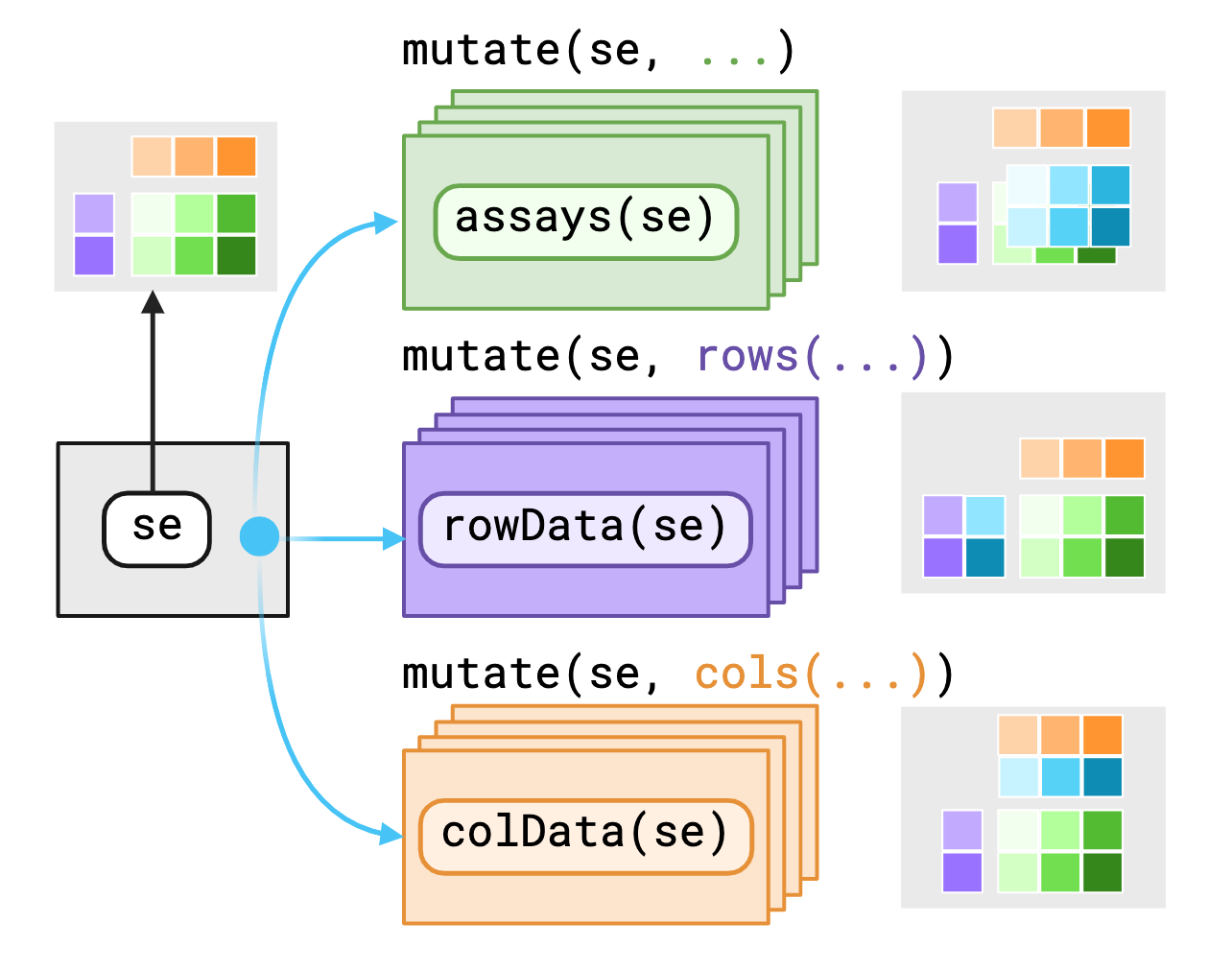
biocmask provides variables as-is to data within their current contexts enabling you to call S4 methods on S4 objects with dplyr verbs. If you require access to variables outside the context, you may use pronouns made available through biocmask to specify where to find those variables.
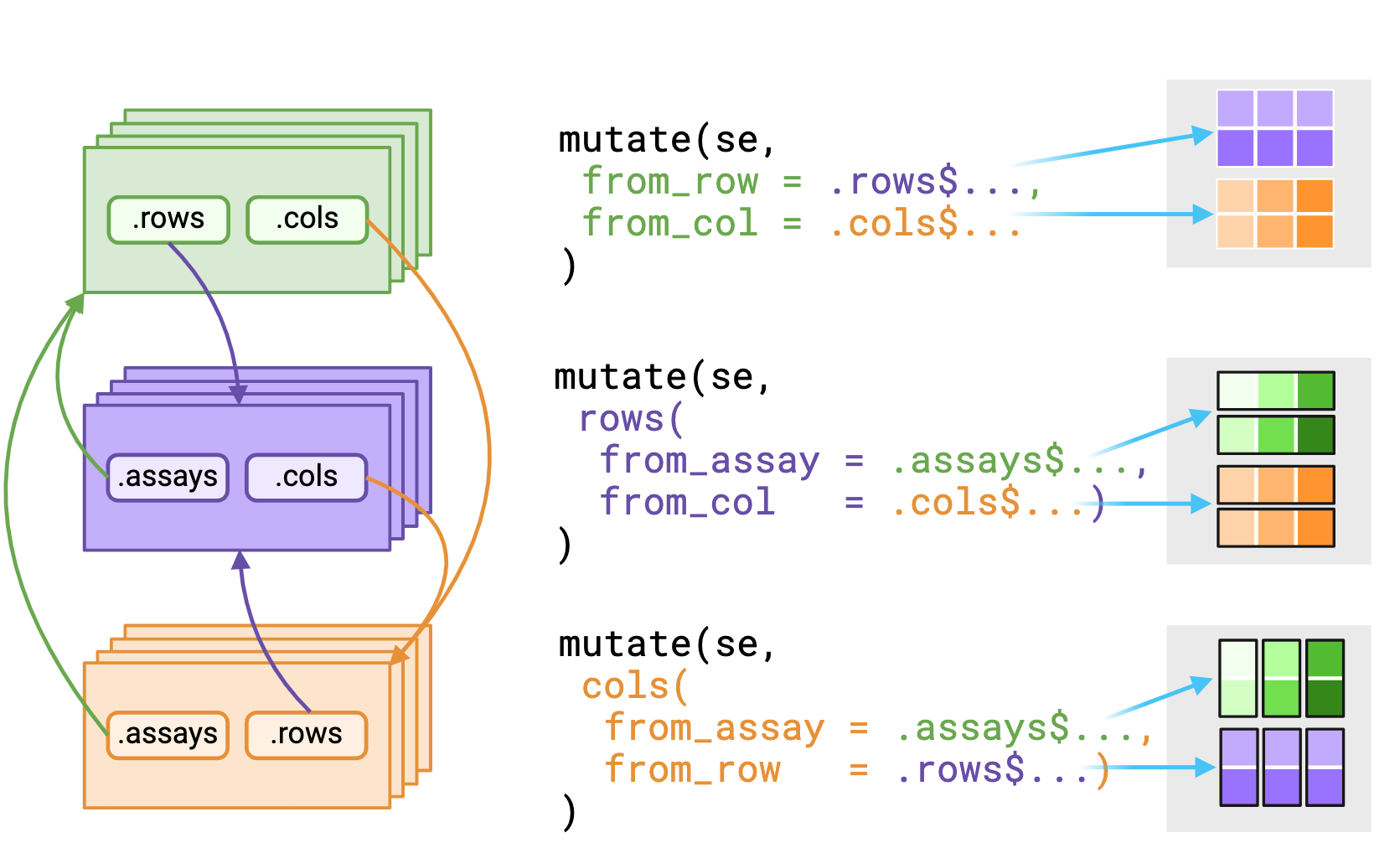
_asis variant that returns underlying data without reshaping it to fit the context. Figure made with Biorender
The .assays, .rows and .cols pronouns outputs depends on the evaluating context. Users should expect that the underlying data returned from .rows or .cols pronouns in the assays context is a vector, replicated to match size of the assay context.
Alternatively, using a pronoun in either the rows() or cols() contexts will likely return a list equal in length to either nrows(rowData()) or nrows(colData()).
Feedback
We would love to hear your feedback. Please post to Bioconductor support site or the #tidiness_in_bioc Slack channel on community-bioc for software usage help, or post an Issue on GitHub, for software development questions.