
Description
plyxp proposes an expressive grammar for manipulating annotated matrix data, with syntax to access, modify, and append matrix data and tabular row and column metadata, including row-wise or column-wise grouped operations. By defining multiple contexts and providing pronouns for specific recall and assignment within and across these contexts, plyxp makes using common dplyr functions as natural as working with a data.frame or tibble.
plyxp is an implementation of this grammar for the R/Bioconductor ecosystem, with efficient abstractions for the SummarizedExperiment class. Data within the SummarizedExperiment are lazily bound to a series of environments, meaning expressions are evaluated only when the user forces their symbols. This gives users more freedom in how they choose to work with their data. plyxp uses data-masking from the rlang package to connect dplyr verbs to SummarizedExperiment slots in an intuitive and unambiguous manner.
The tidySummarizedExperiment package, released with Bioconductor 3.12 in 2020, also provides dplyr-like access to SummarizedExperiment objects within the tidyomics project, allowing datasets to be directly piped into ggplot2 plotting functions, for example. plyxp and tidySummarizedExperiment can be used in parallel, as users engage plyxp functions by casting their SE objects with new_plyxp().
Installing plyxp
# plyxp is available via BiocManager
BiocManager::install("plyxp")
# To use the latest updated version please use the github
remotes::install_github("jtlandis/plyxp")Documentation
See Get started for the package vignette, and Reference for function man pages.
Citation
If you use plyxp in published research, please cite:
Landis JT, Love MI (2026). “Efficient and Tidy Manipulation of Annotated Matrix Data with plyxp.” bioRxiv. 10.64898/2026.05.06.721669
Feedback
We would love to hear your feedback. Please post to Bioconductor support site or the #tidiness_in_bioc Slack channel on community-bioc for software usage help, or post an Issue on GitHub, for software development questions.
How plyxp works
Data masking a SummarizedExperiment
The SummarizedExperiment object contains three main components/“contexts” that we mask, the assays(), rowData()1 and colData().
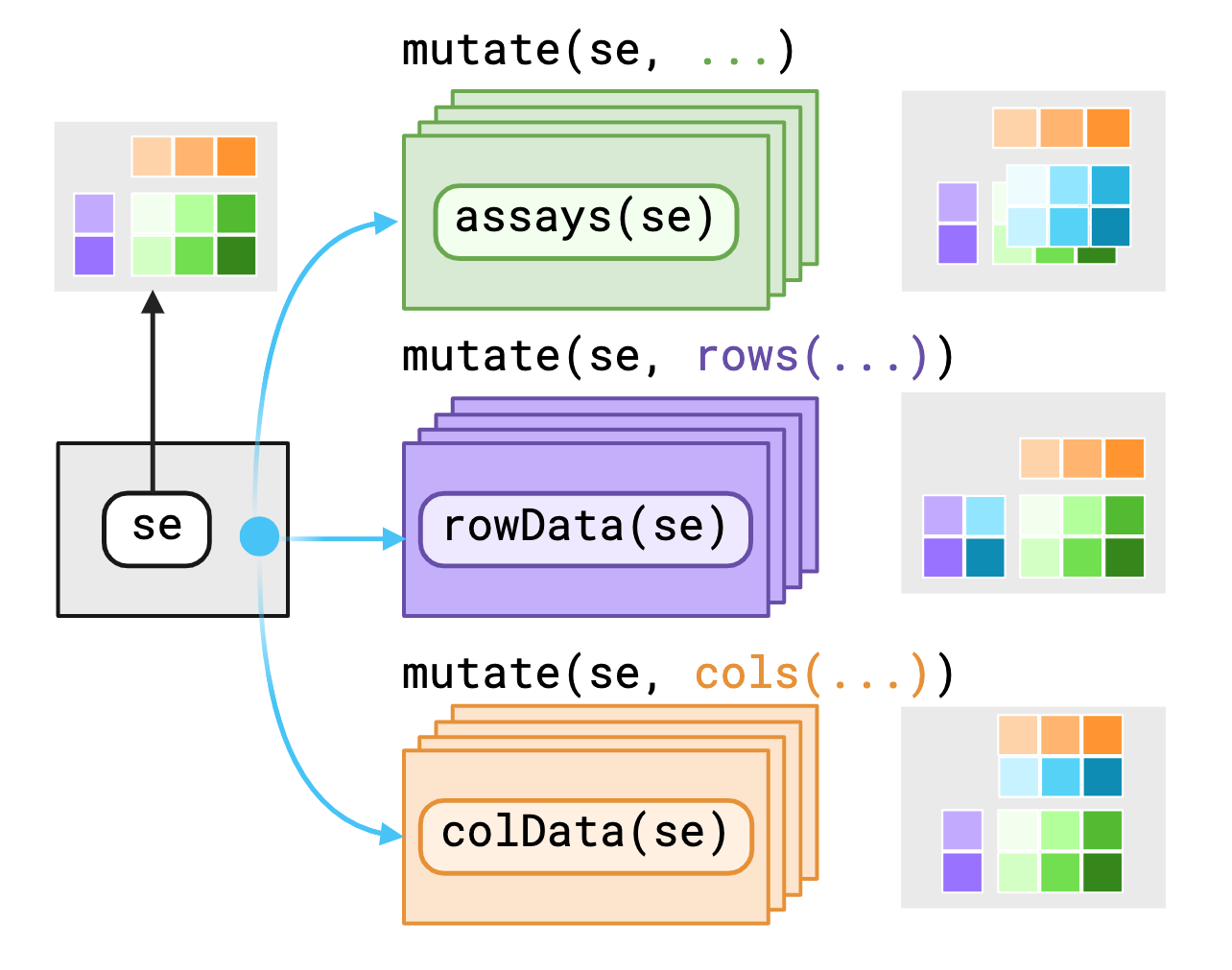
plyxp provides variables as-is to data within their current contexts enabling you to call S4 methods on S4 objects with dplyr verbs. If you require access to variables outside the context, you may use pronouns made available through plyxp to specify where to find those variables.
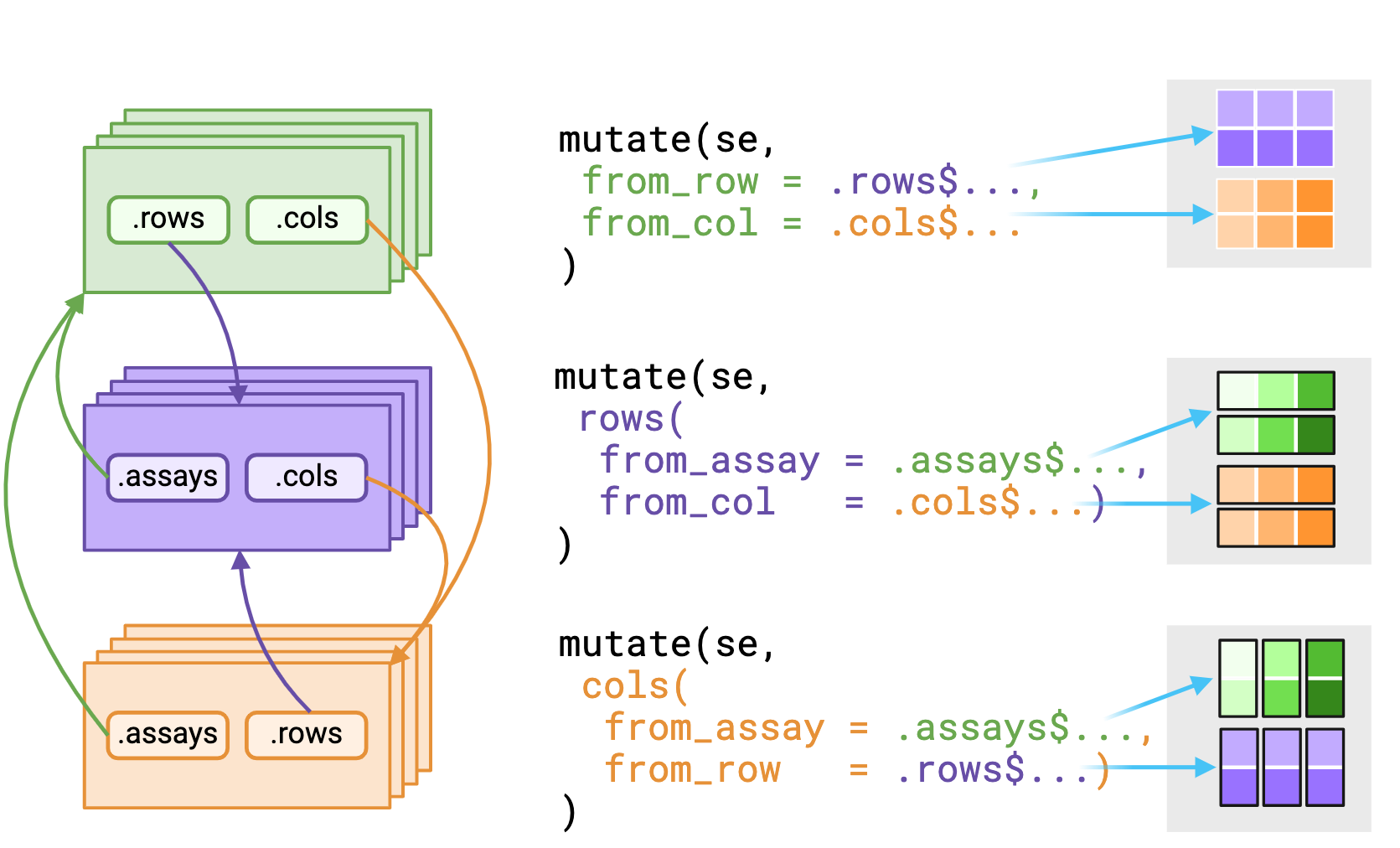
_asis variant that returns underlying data without reshaping it to fit the context. Figure made with Biorender
The .assays, .rows and .cols pronouns outputs depends on the evaluating context. Users should expect that the underlying data returned from .rows or .cols pronouns in the assays context is a vector, replicated to match size of the assay context.
Alternatively, using a pronoun in either the rows() or cols() contexts will likely return a list equal in length to either nrows(rowData()) or nrows(colData()) respectively.
Note on plyxp for Bioc 3.21 or 3.20
plyxp is still under active development. We have recently discovered an error in group_by(xp, rows(foo)) |> summarize(some_assay = <expr>) operations in which the resulting assay matrix was being collected incorrectly. This has been fixed with this commit and has been pushed to plyxp 1.4.3 on Bioconductor version 3.22. With this being said, we cannot update older version of plyxp on Bioconductor 3.21 and 3.20 - however we have cherry-picked this commit into the github branch images.
Thus if you wish to use plyxp from Bioconductor 3.21 or 3.20, please install from github to ensure you have the latest fixes.
remotes::install_github("jtlandis/plyxp@RELEASE_3_21")